$dft (cross-references are
given).
The user normally has to choose only the functional and the grid size, see below. All other parameters have proven defaults.
![[*]](crossref.png) and for definition of the functionals the section
6.2 on page .
and for definition of the functionals the section
6.2 on page .
Example (default input):
$dft functional b-p
). Default is gridsize m3.
Example:
$dft gridsize m3
Possible values for gridtype are
1,…, 6, but gridtype
4 to 6 is only for the use with functional lhf (see
page ). For the definition of gridtype 1 -
3, please refer to Eq. (16), (17) and, (19) in Ref.
[131].
Example (default value):
$dft gridtype 3
Example:
$dft nkk 3

When gridsize 9 is given, you have to specify both, ntheta and nphi (see below), otherwise the program will crash. The restriction for user-defined Lobatto grids is the number of grid points, which must not exceed 2000 grid points.
Example:
$dft gridsize 9 ntheta 30 nphi 60
Example:
$dft old_RbCs_xi
| number of radial grid points = ioffrad + ( |
ioffrad is atom-dependent, the more shells of electrons, the larger ioffrad:
| elements | ioffrad | elements | ioffrad | |
| for H-He | 20 | for K-Kr | 40 | |
| for Li-Ne | 25 | for Rb-Xe | 45 | |
| for Na-Ar | 30 | for Cs-Lw | 50 |
The radial grid size increases further for finer grids:
| gridsize | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||
| radsize | 1 | 2 | 3 | 6 | 8 | 10 | 14 | 9 | 3 |
If you want to converge results with respect to radial grid size, increase radsize in steps of 5, which is convenient (see equation above).
radsize
![]() radsize + incr
radsize + incr
ξ
![]() ξ * scal
ξ * scal
| diffuse integer | 1 | 2 | 3 | 4 | 5 | 6 | ||
| incr | 1 | 2 | 3 | 4 | 5 | 6 | ||
| scal | 1.5 | 2.0 | 2.8 | 4.0 | 5.0 | 6.0 |
For information about the linear scaling parameter ξ, see Eq. (16)-(19) and Table 1 in Ref. [131].
In addition, the reduction of spherical grid points near nuclei
is supressed, i.e. fullshell on is set (see page
).
Note: the keyword radsize integer overrules the setting of
incr; for more information, see p. .
Recommendation: For diffuse cases use gridsize m4 (or larger) in combination with diffuse 2 and check the number of electrons; for more difficult cases use diffuse 4. In case of doubt, verify the calculation with a larger grid, i.e. gridsize 7.
The test suite example
$TURBODIR/TURBOTEST/dscf/short/H3PO4.DSCF.DIFFUSE provides an example of usage;
this also gives reasonable values for damping and orbitalshift to reach
convergence in this and similar cases, see $scfdamp and
$scforbitalshift (p. and p.
).
Example (Recommendation):
$dft gridsize m4 diffuse 2

$dft rhostart 50 rhostop 200one performs a numerical integration for the density and the exchange correlation term for ξ = 0.5,(0.01), 2.0 for given MOs and functional. NOTE: only molecules with a single atom type can be used. The results serve to establish stable, optimal ξ values, see Figure 1 in Ref. [131]. Program stops after this testing.
gridsize 7 radsize 14 fullshell 1 dgrenze 16 fgrenze 16 qgrenze 16 fcut 16Please refer to the corresponding sub-keywords for explanation.
If you want to use the reference grid, you have to skip the keyword gridsize, and type instead reference. Example:
$dft functional b-p reference
.
Example for the usage of fullshell:
$dft functional b-p gridsize m4 fullshell
 ).
.
).
.
Example for switching off gridordering:
$dft gridordering 0
Ex, Ey, Ez or by x, y, z, |E|.
See also $fldopt.
Example:
$electrostatic field
0.1000E-03 0.000 0.000
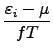
Calculation of the fractional occupation numbers starts when the current HOMO-LUMO gap drops below the value given by hlcrit (default: 0.1). The initial temperature given by tmstrt (default: 300K) is reduced at each SCF cycle by the factor tmfac (default: 1.0) until it reaches the value tmend (default: 300K). Note that the default values lead to occupation numbers calculated at a constant T = 300K. Current occupation numbers are frozen if the energy change drops below the value given by stop (default: 1 ⋅10-3). This prevents oscillations at the end of the SCF procedure.
Calculation of fractional occupation numbers often requires much higher damping and orbital shifting. Please adjust the values for $scfdamp and $scforbitalshift if you encounter convergence problems.
In UHF runs this option can be used to automatically locate the lowest spin state. In order to obtain integer occupation numbers tmend should be set to relatively low value, e.g. 50K.
Calculation of fractional occupation numbers should be used only for single point calculations. When used during structure optimizations it may lead to energy oscillations.
The optional nue value (number of unpaired electrons) can be used to force a certain multiplicity in case of an unrestricted calculation. nue=0 is for singlet, nue=1 for dublet, etc.
$scfmo. This enables to evaluate
properties or perform geometry optimizations
in the presence of an external field.
Caution: don't use the RI approximation for all these calculations since this will lead to non-negligible errors!!
The following condition must be met:
and rhfshells 1 or 2.
It is advisable to set $thize as small as possible
(e.g. $thize 0.1d-08) and to remove the keyword
$scfdump.
Note: this keyword does not work for parallel runs.
nirreps will hold the total number of displayed orbitals after the successful run.
Example:
$mo output format(3(2x,d15.8))
a 1-5 ( 2.00000000000000 ) a 6 ( 1.99949836999366 ) a 7 ( 1.99687490286069 ) a 8 ( 1.00000000000000 ) a 9 ( .00312509713931 ) a 10 ( .00050163000634 )
<x> <y> <z> <q>with
, , being the coordinates and
its charge,e.g.
$point_charges thr=<real> self-energy list
2. 2. 2. 5.
5. 0. 0. 2.5
In addition the following optional arguments may be given:
- thr
- distance threshold for discarding redundant point charges,
default value 10-6.
- selfenergy
- if given, the selfenergy of the point charge array
will
will be included in the energy and the gradient
- list
- print all point charges in the output (default is to
print
the point charges only if less than 100 charges given)
- $prediag
-
concerns the first SCF iteration cycle if start MOs from an EHT guess
are used.
The SCF iteration procedure requires control mechanisms to ensure
(fast) convergence, in TURBOMOLE these are based on orbital energies
εi of the preceeding iteration used for level shifting and
damping (besides DIIS, see below). This feature cannot be used in the
first iteration if EHT MOs are employed as start, since
εi
are not available. The keyword $prediag provides
'
εi of the zeroth iteration' by diagonalization of occ-occ
and virt-virt part of the first Fock matrix, to allow for level
shifting etc..
See $scfdiis below.
- $restart dscf twoint
-
Try a dscf restart. The program will read the data group
$restartd (which must exist, also $scfmo has to exist!) and
continue the calculation at the point where it ended before. If the additional
option twoint is appended, the program will read the two-electron
integrals from the files specified in $scfintunit, so there will be
almost no loss of cpu-time.
All this information is normally provided by the previous dscf run if the
keyword $scfdump (see there) was given.
- $restartd data
-
Data provided by a previous dscf run that has been interrupted. This keyword is
created when $scfdump was given.
- $rundimensions data
-
is set by define so usually no changes are necessary. The
dimensions must be greater or equal to those actually required,
i.e. you can delete basis functions and keep rundimensions. This
keyword is not necessary for small cases.
Example:
dim(fock,dens)=6072
natoms=6
nshell=34
nbf(CAO)=108
nbf(AO)=98
dim(trafo[SAO<-->AO/CAO])=256
rhfshells=1
- $scfconv integer
-
SCF convergency criterion will be
10-integer for the energy.
Gradients will only be evaluated if integer > 6.
- $scfdamp start=<.500> step=<.050> min=<.100>
-
Damping parameters for SCF iterations in order to reduce oscillations.
The old Fock-operator is added to the current one with weight
0.5 as start; if convergence is good, this weight is then
reduced by the step 0.05 in each successive iteration
until the minimum of 0.1 is reached.
(These are the default settings of define for closed-shell RHF).
DSCF automatically tries to adjust the weight to optimize
convergence but in difficult cases it is recommended to
start with a large weight, e.g. 1.5, and to set the minimum
to a larger value, e.g. 0.5.
- $scfdebug options
-
Flags for debugging purposes. Following options are available:
- vectors integer
-
Output level concerning molecular orbitals. integer=0 (default) means
minimal output,
>1 will output all start MOs and all MOs in each iteration.
- density integer
-
Output level concerning difference density matrices.
- debug integer
-
integer > 0 will dump a lot of information--be careful!
- $scfdenapproxl integer
-
Direct SCF procedures build the Fock matrix by exploiting
information from previous iterations for better efficiency. With
this keyword information from the last integer iterations
will be used. This feature is switched on with the default value 20,
even if the keyword is absent. The user may reduce the number of
iterations employed to smaller values (e.g. 10) in cases were
numerical stability could become an issue. With the value 0 this
feature is switched off; the Fock matrix is constructed from scratch
in each iteration.
- $scfdiis options
-
Control block for convergence acceleration via Pulay's DIIS
 .
.
Options are:
- errvec=char
- specifies the
kind of error vector to be used (two different kind of DIIS
algorithms)
- char='FDS' or 'SDF' or
'FDS-SDF'
-
uses
(FDS - SDF) as error vector.
- char= none
-
no DIIS
- char= sFDs
-
use
S-1/2FDS1/2 - transposed
Further suboptions:
- maxiter=integer
-
maximum number of iterations used for extrapolation.
- debug=integer
-
debug level (default: 0)
- integer=1
- print applied DIIS coefficients
- integer=2
- print DIIS matrix and
eigenvalues, too
- qscal=real
-
scaling factor in DIIS procedure: qscal > 1 implies
some damping, qscal = 1.0: straight DIIS.
- thrd=real
-
directs the reduction of qscal to qscal = 1.0 (no damping in
DIIS), done if
|| errvec|| < thrd.
Defaults for $prediag (see above) and $scfdiis
errvec=FDS-SDF, maxiter=5, qscal=1.2,
thrd=0.0, this implies DIIS damping in all iterations,
prediag is switched of.
Recommended:
errvec=sFDs leads to the following defaults:
qscal=1.2, for SCF runs: maxiter=6 and
thrd=0.3, prediag is off; for DFT runs: maxiter=5
and thrd=0.1 prediag is on. If you want to switch off prediag
put
$prediag none.
- $scfdump
-
Dump SCF restart information onto data group $restartd and
dump SCF MOs in each iteration onto $scfmo (scfdump = iter).
Additionally, a data block $scfiterinfo will be dumped
containing accumulated SCF total-, one- and two-electron energies of
all previous SCF iterations. Information that will allow you to
perform a restart if your calculation aborts will be dumped on
data group $restartd (see also $restart).
- $scfintunit options
-
Disc space specification for two-electron integrals. The following suboptions
are available (and necessary):
- unit=integer
-
Fortran unit number for this file. Unit numbers 30,31,... are recommended.
- size=integer
-
Filespace in megabytes for this file. size=0 leads to a fully direct
run.
size is set by a statistics run, see $statistics.
DSCF switches to direct mode if the file space is exhausted.
- file=char
-
Filename. This may also be a complete path name, if you want to store the
integrals in a special directory. Make sure the file is local,
otherwise integrals are transmitted over the network.
Thus your data group $scfintunit may look like this:
$scfintunit
unit=30 size=35 file=twoint1
unit=31 size=35 file=/users/work/twoint2
Maximal 30 files may be specified in this way.
- $scfiterlimit integer
-
Maximum number of SCF iterations (default: 30).
- $scfmo none file=char
-
Input/output data group for SCF MOs. You can specify
- none
-
To perform a calculation without a start vector (i.e. use a core Hamiltonian
guess).
- file=char
-
The file where the MOs are written on output (default: mos).
These two options can also be used for $uhfmo_alpha and
$uhfmo_beta to
use a core guess and write the molecular orbitals to file.
After running define or a TURBOMOLE calculation additional options may
appear specifying the origin of the MOs:
- expanded
-
These MOs were obtained by projection form another basis set. They should not
be used for wavefunction analysis.
- scfconv=integer
-
The MOs are converged SCF MOs , the convergence criterion applied was
10-integer
- scfdump=integer
-
The MOs are unconverged SCF MOs which were written on this data group after
iteration
integer. The latter three options are mutually exclusive.
- format(format string)
-
This specifies the FORTRAN format specification which was used for MO output.
The standard format
is (4d20.14). (See data group $mo output format.)
Example:
Your data group $scfmo could look like this after a successful
TURBOMOLE run :
$scfmo scfconv=7 format(3(1x,d19.13))
1 a1 eigenvalue=-.524127 nsao=6
.1234567890123d+01 -.1234567890123d+00 .1234567890123d-01
.1234567890123d+01 -.1234567890123d+00
3 a2 eigenvalue=-.234810
...
- $scforbitalorder on/off
-
Order SCF MOs with respect to their energies (default: on)
- $scforbitalshift options
-
To assist convergence, either the energies of unoccupied MOs can be shifted
to higher energies or, in open-shell cases, the energies of closed-shell MOs
to lower energies.
In general a large shift may help to get better convergence.
Options are:
- noautomatic
-
Automatic virtual shell shift switched off.
- automatic real
-
Automatic virtual shell shift switched on; the energies of virtual orbitals
will be shifted if the HOMO-LUMO gap drops below real such that a
gap of real is sustained.
This is the default setting if the keyword is missing with real=0.1.
- closedshell=real
-
Option for open-shell cases. Closed shells are shifted to lower energies by
real. The default shift value is closedshell=0.4.
Note: Normally this will disable the automatic shift of energies of
virtual
orbitals! To override this, you should append an exclamation mark to the
'automatic' switch, i.e. specify 'automatic! real'.
- individual
-
Set shifts for special occupied MOs. To use this option, start the line with
the symmetry label and the list of MOs within this symmetry and append the
desired shift in brackets as in the following example:
a1 1,2,4-6 (-.34)
b1 8 (+.3)
- $scftol real
-
Integral evaluation threshold. Integrals smaller than real will
not be evaluated. Note that this threshold may affect accuracy and the
convergence properties if it is chosen too large.
If $scftol is absent, a default value will be taken obtained from
$scfconv by
real = 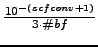 (#bf =
number of basisfunctions).
(#bf =
number of basisfunctions).
- $scratch files
-
The scratch files allocated by dscf can be placed anywhere
in your file systems instead of the working directory by referencing
their pathnames in this data group. All possible scratch files are listed
in the following example:
$scratch files
dscf dens path1/file1
dscf fock path2/file2
dscf dfock path3/file3
dscf ddens path4/file4
dscf statistics path7/file7
dscf errvec path8/file8
dscf oldfock path9/file9
dscf oneint path10/file10
The first column specifies the program type (dscf stands for SCF
energy calculations, i.e. the dscf program), the
second column the scratch file needed by this program and the third column
the pathname of the file to be used as scratch file.
- $statistics options
-
The following options are allowed
- off
- Do not perform integrals statistics
- dscf
- Perform integrals statistics for dscf
- kora
- see KORA
- mpgrad
- see mpgrad
- polly
- see POLLY
- dscf parallel
- see PARALLEL PROCESSING
Options kora, dscf parallel, grad, mpgrad, polly will be described
in the related chapters.
If $statistics dscf has been given integral prescreening will be
performed (which is an n4-step and may therefore be time-consuming) and
a table of the number of stored integrals as a function of the two parameters
$thize and $thime will be dumped.
Afterwards, the filespace needed for the current combination of $thize
and
$thime will be written to the data group ($scfintunit)
and $statistics dscf will be replaced by $statistics off.
- $thime integer
-
Integral storage parameter, which is related to the time needed to calculate
the integral. The larger integer the less integrals will be stored.
The default value is integer = 5. (see also $thize,
$statistics)
- $thize real
-
Integral storage parameter, that determines, together with $thime, the
number of integrals stored on disc. Only integrals larger than real
will be stored. The default value is real = 0.100E-04.