



Next: Keywords for Module Relax
Up: Format of Keywords and
Previous: RIMP2: Essential Keywords
Contents
Index
Keywords for Module RICC2
Note that beside the keywords listed below the outcome of the
ricc2 program also depends on the settings of most thresholds that
influence the integral screening
(e.g. $denconv, $scfconv, $scftol)
and for the solution of Z vector
equation with 4-index integrals
(for relaxed properties and gradients)
on the settings for integrals storage
in semi-direct SCF runs
(i.e. $thime, $thize, $scfintunit).
For the explanation of these keywords see Section 15.2.5.
- $cbas file=auxbasis
-
Auxiliary basis set for RI approximation.
For details Section 15.2.12.
- $freeze
-
Freeze orbitals in the calculation of correlation and excitation energies.
For details see Section 15.2.12.
- $printlevel 1
-
Print level. The default value is 1.
- $tmpdir /work/thisjob
-
Specify a directory for large intermediate files (typically three-index
coulomb
integrals and similar intermediates), which is different from the
directory
where the ricc2 program is started.
- $maxcor 20
-
The data group $maxcor adjusts the maximum size of core
memory in MB which will be allocated during the RI-CC2 run.
This keyword can be set in define or with the Rimp2prep
tool, the default is 20MB.
$maxcor has a large influence on computation times for RI-CC2 runs!
It is recommended to set $maxcor to ca. 75-85% of the available
(physical) core memory.
- $spectrum unit
-
The calculated excitation energies and corresponding oscillator
strengths are appended to a file named 'spectrum'. Possible values
of unit are eV, nm and cm-1 or rcm. If no unit is
specified, excitation energies are given in a.u.
- $cdspectrum unit
-
The calculated excitation energies and corresponding rotatory
strengths are appended to a file named
'cdspectrum'. unit can have the same values as in
$spectrum.
- $laplace
conv = 5
The purpose of this data group is twofold: It activates the
Laplace-transformed implementation of SOS-MP2 in the ricc2 module
(if the sos option has been specified in $ricc2)
and it provides the options to specify the technical details for
the numerical Laplace-transformation.
- conv
-
Threshold for the numerical integration used for the Laplace transformation
of orbital energy denominators.
The grid points for the numerical integration are determined such that
is the remaining root mean squared error (RMSE) of the Laplace transformation
is
< 10-conv.
By default the threshold is set to the value of conv
given in $ricc2 (see below).
- $ricc2
ccs
cis
mp2 d1diag
cis(d) energy only
cis(dinf)
adc(2)
cc2
restart
norestart
hard_restart
nohard_restart
conv = 8
oconv = 7
lindep = 15
maxiter = 25
mxdiis = 10
maxred = 100
iprint = 1
fmtprop = f15.8
geoopt model=cc2 state=(a" 2)
scs cos=1.2d0 css=0.3333d0
sos
gsonly
d1diag
specifies the ab initio models (methods) for ground and
excited states and the most important parameters and thresholds
for the solution of the cluster equations, linear response equations
or eigenvalue problems.
If more than one model is given, the corresponding calculations
are performed successively. Note: The CCS ground state energy
is identical with the SCF reference energy, CCS excitation energies
are identical to CIS excitation energies. The MP2 results
is equivalent to the result from the rimp2 module.
cis(dinf) denotes the iterative CIS(D) variant CIS(D∞).
- mp2 d1diag
-
Request the calculation of the D1 diagnostic in MP2 energy calculations
(ignored in MP2 gradient calculations). Note that the evaluation of the
D1 diagnostic increases the computational costs of the RI-MP2 energy
calculation roughly by a factor of 3.
- cis(d) energy only
-
If the energy only flag is given after the cis(d) keyword, it is
assumed that only excitation energies are requested. This
switches on some
shortcuts to avoid the computation of intermediates needed e.g. for the
generation of
improved start vectors for CC2.
- (no)restart
-
If the restart flag is set, the program will
try to restart the CC2 calculations from previous solution vectors on
file. If the norestart flag is set no restart will be done. Default is to do a
restart for CC2 if and
only if the file CCR0--1--1---0 exists. Note: There is
no restart
possibility for CCS/CIS or MP2/CIS(D).
- (no)hard_restart
-
If the hard_restart flag is set, the
program will try to reuse integrals and intermediates from a previous
calculation. This
requires that the restart.cc file has been kept, which
contains check sums and some
other informations needed. The hard_restart flag is
switched on by default, if
the restart.cc file is present.
- conv
- The conv parameter gives the
convergence threshold for
the CC2 ground state energy as
10-conv. The default value
is taken from the
data group $deneps.
- oconv
-
The oconv parameter gives an additional threshold for
the residual of the cluster equations (vector function). If this
parameter is given, the
iterations for the cluster equations are not stopped before the norm of
the residual is
< 10-oconv. By default the threshold is set to oconv = conv-1, or 10× deneps if no input for
conv is given.
- lindep
-
If the norm of a vector is smaller than
10-lindep,
the vector is assumed to be zero. This threshold is also used to test
if a set of vectors
is linear dependent. The default threshold is 10-15.
- maxiter
-
gives the maximum number of iterations for the solution of the
cluster equations, eigenvalue problems or response equations (default:
25).
- mxdiis
-
is the maximum number of vectors used in the DIIS procedures for
CC2 ground state or excitation energies (default: 10).
- maxred
-
the maximum dimension of the reduced space in the solution of
linear equations (default: 100).
- iprint
-
print level, by default set to 1 or (if given) the the value of
the $printlevel data group.
- fmtprop
-
Fortran print format used to print several results
(in particular one-electron properties and transition moments) to
standard output.
- geoopt
-
specify wavefunction and electronic state for which a geometry
optimization is intended. For
this model the gradient will be calculated and the energy and gradient
will be written onto
the data groups $energy and $grad. Required for geometry
optimizations using the jobex script.
Note, that in the present version gradients are only available
for ground states at the MP2 and CC2 and for excited states at the
CC2 level
and not for ROHF based open-shell calculations.
Not set by default. The default model is CC2, the default electronic
state
the ground state. To obtain gradients for the lowest excited state
(of those
included in the excitation energy calculation, but else of
arbitrary
multiplicity and symmetry) the short cut s1 can be used.
x is treated as synonym for the ground state.
- scs
-
the opposite-spin scaling factor cos and the
same-spin scaling factor css can be choosen.
If scs is set without further input,
the SCS parameters cos=6/5 and css=1/3 are applied.
This keyword can presently only be used in connection with MP2.
- sos
-
the SOS parameters cos=1.3 and css=0.0 are applied.
This keyword can presently only be used in connection with MP2.
- d1diag
-
request the calculation of the D1 diagnostic for the ground
state wavefunction. Only needed for MP2 (see above for the
alternative input option mp2 d1diag).
For all other correlated
methods the D1 diagnostic is evaluated by default
(without significant extra costs).
- $rir12
ansatz
r12model
comaprox
cabs
examp
pairenergy
local
corrfac
cabsingles
ump2fixed
- ansatz char
-
char=1, 2* or 2
The ansatz flag determines which ansatz is
used to calculate the RI-MP2-F12 ground state energy.
(Ansatz 2 is used if ansatz is absent.)
- r12model char
-
char=A, A' or B
The r12model flag determines which approximation model
is used to calculate the RI-MP2-F12 ground state energy.
(Ansatz B is used if r12model is absent.)
- comaprox char
-
char=F+K or T+V
The comaprox flag determines the method used
to approximate the commutator integrals
[T, f12].
(Approximation T+V is used if comaprox is absent.)
- cabs char val
-
char=svd or cho
The cabs flag determines the method used
to orthogonalize the orbials of the CABS basis. val is
the threshold below which CABS orbitals are removed from the
calculation.
(svd 1.0d-08 is used if cabs is absent.)
- examp char
-
char=noinv, fixed or inv
The examp flag determines which methods are
used to determine the F12 amplitudes. For inv the
amplitudes are optimised using the orbital-invariant method.
For fixed and noinv only the diagonal amplitudes
are non-zero and are either predetermined using the coalescence
conditions (fixed), or optimised (noinv--not orbital
invariant). If char=inv, the F12 energy contribution
is computed using all three methods.
(The fixed method is used if examp is absent.)
- pairenergy char
-
char=off or on
If char=off (default), the print out of the F12 contribution
to the pair energies is supressed. The summary of the RI-MP2-F12
correlation energies is always printed out.
- local char
-
char=off, boys or pipek
The active occupied molecular orbitals are localized by Boys or
Pipek-Mezey method. Currently, the local flag is restricted
to closed shell cases within approximation A and the linear correlation
factor. If local is absent, no localization is performed.
- corrfac char
-
char=LCG or R12
The corrfac flag determines which correlation factor is
used for the geminal basis. LCG requires the data group
$lcg, which contains the information regarding exponents and
coefficients of the linear combination of Gaussians.
- cabsingles char
-
char=off or on
The cabsingles flag determines whether or not the
single excitations into the CABS basis are computed.
The CABS singles are computed in any case if the CABS Fock
matrix elements are computed anyway
for the F12 calculation (i.e.,
for ansatz 2 or r12model B or comaprox F+K).
- ump2fixed char
-
char=diag or full
The umpfixed flag controls which fixed-amplitude method
is used for calculations using ROHF or UHF references. full
is more computationally demanding than diag, but gives
energies closer to the inv method.
If ump2fixed is absent, the full method is used.
- $excitations
-
irrep=au multiplicity=1 nexc=4 npre=6 nstart=8
irrep=bg multiplicity=3 nexc=2 npre=4 nstart=5
spectrum states=all operators=diplen,dipvel
exprop states=all operators=qudlen
xgrad states=(ag{3} 1)
conv = 6
thrdiis = 2
preopt = 3
leftopt
bothsides
In this data group you have to give additional input for calculations
on excited states:
- irrep
-
the irreducible representation.
- multiplicity
-
spin multiplicity (1 for
singlet, 3 for triplet);
default: singlet, not needed for UHF.
- nexc
- the number of excited states to be
calculated within this irrep
and for this multiplicity.
- npre
- the number of roots used in
preoptimization steps
(default:
npre = nexc).
- nstart
-
the number of start vectors
generated or read from file
(default: nstart = npre).
- spectrum
-
This flag switches on the calculation of oscillator strengths
for
excited state--ground state transitions. Setting the parameter
states=all
is mandatory for the calculation of transition properties in
the present version.
The operators flag can be followed by a list of
operators (see below) for which the
transition properties will be calculated. Default is to compute
the oscillator
strengths for all components of the dipole operator.
- exprop
-
require calculation of
first-order properties for excited states.
For the states option see spectrum option above;
for details for the operators input see below.
- xgrad
-
request calculation of the gradient for the
total energy of an excited state. If no state is specified, the
gradient
will be calculated for the lowest excited state included in the
calculation
of excitation energies (Note that only a single state should be
specified; simultaneous calculation of gradients for several
states is
in the present version not possible.).
- conv
- convergence threshold for norm of
residual
vectors in eigenvalue problems is set to
10-
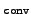 .
If not given, a default value is used, which is
chosen as
max(10-, 10-
.
If not given, a default value is used, which is
chosen as
max(10-, 10-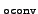 , 10-6),
, 10-6),
where conv refers to the values given in
the data group $ricc2.
- preopt
-
convergence threshold used for
preoptimization
of CC2 eigenvectors is set to
10-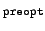 (default:
3).
(default:
3).
- thrdiis
-
threshold (
10-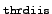 )
for residual norm below which DIIS
extrapolation is switched on in the modified Davidson algorithm for
the non-linear CC2 eigenvalue problem (default: 2).
)
for residual norm below which DIIS
extrapolation is switched on in the modified Davidson algorithm for
the non-linear CC2 eigenvalue problem (default: 2).
- leftopt
-
If the flag leftopt is set the left eigenvectors are computed
(default is to compute the right eigenvectors, for test purposes only).
- bothsides
-
The bothsides flag enforces the calculation of both, the left and
the right eigenvectors (for test purposes only).
- $response
fop unrelaxed_only operators=diplen
gradient
conv = 6
zconv = 6
semicano
nosemicano
thrsemi = 3
In this data group you have to give additional input for the calculation
of ground state properties and the solution of response equations:
- fop
- This flag switches on the calculation of
ground state first-order
properties (expectation values).
The operators flag can be followed by a list of operators
(see below)
for which the first-order properties will be calculated. Default is to
compute
the components of the dipole and the quadrupole moment.
The option unrelaxed_only suppress the calculation of
orbital-relaxed first-order
properties, which require solution the CPHF-like Z-vector equations.
Default is the calculation of unrelaxed and orbital-relaxed first-order
properties.
The unrelaxed_only option will be ignored, if the
calculation
of gradients is requested (see gradient option below and
geoopt in data group $ricc2).
- gradient
-
require calculation of geometric gradients.
In difference to the geoopt keyword in the data group
$ricc2
this can be used to compute gradients for several methods
within a loop over models; but gradients and energies will not be
written to the data groups $grad and $energy as needed
for geometry optimizations.
Note, that in the present version gradients are only available for
MP2 and CC2 and only for a closed-shell RHF reference.
- conv
- convergence threshold for norm of residual
vectors in linear response equations is set to
10-.
If not given in the $response data group,
a default value is used, which is
chosen as
max(10-,
10-, 10-6),
where conv and oconv refer to the values
given in the data group $ricc2.
- zconv
-
convergence threshold for the norm of the
residual vector in the solution of the Z vector equations
will be set to
10-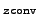 .
.
- semicano
-
use semi-canonical formulation for the
calculation of (transition) one-electron densities. Switched on by default.
The semi-canonical formulation is usually computationally more efficient
than the non-canonical formulation. Exceptions are systems with many
nearly degenerate pairs of occupied orbitals, which have to be treated
in a non-canonical way anyway. (See also explanation for
thrsemi below).
- nosemicano
-
use non-canonical formulation for the
calculation of (transition) one-electron densities. Default is to use
the semi-canonical formulation.
- thrsemi
-
the threshold for the selection of nearly degenerate
pairs of occupied orbitals which (if contributing to the density) have to
be treated in a non-canonical fashion will be set to
10-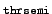 .
If set to tight the semi-canonical algorithm will become inefficient,
if the threshold is to large the algorithm will become numerical unstable
.
If set to tight the semi-canonical algorithm will become inefficient,
if the threshold is to large the algorithm will become numerical unstable
- zpreopt
-
threshold for preoptimizating the so-called Z vector (i.e. the
lagrangian multipliers for orbital coefficients) with a preceding
RI-CPHF calculation with the cbas auxiliary basis. The RI-CPHF
equations will be converged to a residual error
< 10-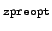 .
Default is
.
Default is zpreopt=4. This preoptimization can reduce significantly
the computational costs for the solution of the Z vector equations for
large basis sets, in particular if they contain diffuse basis functions.
For calculations on large molecules with small or medium sized basis sets
the preoptimization becomes inefficient compared to the large effects
of integral screening for the conventional CPHF equations and should
be disabled.
This option is automatically disabled for ricc2 calculations
based on foregoing RI-JK Hatree-Fock calculation.
- nozpreopt
-
disable the preoptimization of the Z vector by a preceding RI-CPHF
calculation with the cbas basis set.
(Note that the preoptimization is automatically deactivated if the
ricc2 calculation is based on a foregoing RI-JK Hatree-Fock calculation.)
Common options for keywords in the data groups
$ricc2, $response, and $excitations:
- operators=diplen,dipvel
-
input of
operator labels for first-order properties, transition moments, etc.
Currently implemented operators/labels are
- overlap
- overlap (charge) operator: the
integrals evaluated in the AO basis are
〈μ| ν〉
- diplen
- dipole operator in length gauge:
〈μ| riO| ν〉 with i = x, y, z; the index
O indicates dependency on the origin (for expectation values of
charged molecules), which in the present version is
fixed to (0, 0, 0)
(all three components, individual components can be specified
with the labels xdiplen, ydiplen, zdiplen).
- dipvel
- dipole operator in velocity gauge:
〈μ|∇i| ν〉
(all three components, individual components can be specified
with the labels xdipvel, ydipvel, zdipvel).
- qudlen
- quadrupole operator
〈μ| rOirOj| ν〉
(all six components, individual components can be specified with
the labels xxqudlen, xyqudlen, xzqudlen,
yyqudlen, yzqudlen, zzqudlen).
If all six components are present, the program will automatically
give the electronic second moment tensor (which involves only the
electronic contributions) Mij, the isotropic second moment
α = 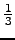 trM and the anisotropy
trM and the anisotropy
β =  . . |
|
Furthermore the traceless quadrupole moment
Θij = 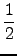 〈3rirj - r2δij〉 〈3rirj - r2δij〉 |
|
(including nuclear contributions) is given.
- angmom
- angular momentum
〈μ| LOi| ν〉
(all three components, individual components can be
specified with the labels
xangmom, yangmom, zangmom).
- nef
- electronic force on nuclei
〈μ|
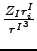 | ν〉, where ZI is the charge of the nucleus
I
and rI is the position vector of the electron relative to the nucleus
(all
three components for all nuclei: the labels are xnef001, ynef001,
znef001, xnef002, etc. where the number depends on the order
in
the coord file).
| ν〉, where ZI is the charge of the nucleus
I
and rI is the position vector of the electron relative to the nucleus
(all
three components for all nuclei: the labels are xnef001, ynef001,
znef001, xnef002, etc. where the number depends on the order
in
the coord file).
- states=all
-
specification of states for which
transition moments or first-order properties are to be calculated. The default
is all, i.e. the calculations will be done for all excited states for
which excitation energies have been calculated. Alternatively, one can select a
subset of these listed in parentheses, e.g.
states=(ag{3} 1,3-5; b1u{1} 1-3; b2u4).
This
will select the triplet ag states no. 1, 3, 4, 5 and the singlet
b1u states
no. 1, 2, 3 and the singlet (which is default if no {} is found)
b2u
state no. 4.
- $D2-diagnostic
-
Calculate the double-substitution-based diagnostics D2.
- $cc2_natocc
-
Write MP2/CC2 natural occupation numbers and natural orbitals
to a file.
- $cgrad 1000
-
Calculate the error functional
δRI for
the RI approximation of (ai| bj) integrals
δRI =
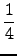
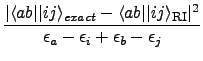
and its gradients with respect to exponents and coefficients
of the auxiliary basis set as specified in the data group $cbas.
The results are written to $egrad scaled by the factor given
with the keyword $cgrad and can be used to optimize auxiliary basis
sets
for RI-MP2 and RI-CC2 calculations (see Section 1.5).
Next: Keywords for Module Relax
Up: Format of Keywords and
Previous: RIMP2: Essential Keywords
Contents
Index
TURBOMOLE