The unrelaxed first-order properties are calculated from the variational
excited states Lagrangian [106], which for the calculation
of unrelaxed properties is decomposed into a ground state contribution and
Lagrange functional for the excitation energy which leads to expressions for
difference densities (or changes of the density matrix upon excitations):
| 〈V〉ur, ex | = | 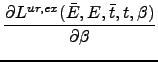 |
(9.20) |
| = | (9.21) |
In the present implementation, the ground-state and the difference
density matrices are evaluated separately.
The calculation of excited-state first-order properties thus
requires also the calculation of the ground-state density matrix.
In addition, the left (
![]() ) and right (Eμ) eigenvectors
and the Lagrangian
multipliers
) and right (Eμ) eigenvectors
and the Lagrangian
multipliers
![]() need to be determined for each excited state.
The disk space and CPU requirements for solving the equations for
need to be determined for each excited state.
The disk space and CPU requirements for solving the equations for
![]() and
and
![]() are about the same as those for the calculation of the
excitation energies. For the construction of the density matrices in
addition some files with
are about the same as those for the calculation of the
excitation energies. For the construction of the density matrices in
addition some files with
![]() (nrootN2) size are written,
where nroot is the number of excited states.
(nrootN2) size are written,
where nroot is the number of excited states.
The single-substitution parts of the excited-states Lagrangian
multipliers
![]() are saved in files named
are saved in files named
CCNE0-s--m-xxx.
For the calculation of first-order properties for excited states,
the keyword exprop must be added with appropriate options to the
data group $excitations; else the input is same as for the
calculation of excitation energies:
$ricc2 cc2 $response fop unrelaxed_only operators=diplen,qudlen $excitations irrep=a1 nexc=2 exprop states=all operators=diplen,qudlenBecause for calculation of excited-states first-order properties also the (unrelaxed) ground-state density is evaluated, it is recommended to specify also ground-state first-order properties in the input, since they are obtained without extra costs.
To obtain orbital-relaxed first-order properties or analytic derivatives
(gradients) the Lagrange functional for the excited state
in Eq. (9.18) is--analogously
to the treatment of ground states--augmented by the equations for
the SCF orbitals and
external perturbations are (formally) included in the SCF step,
i.e. also in the Fock operator.
Since relaxed densities or often computed in connection with geometry
optimizations for individual states (rather than simultaneously for
many states) a Lagrangian for the total energy of the excited state
is used. This has the advantage the only one equation for Lagrangian multipliers
for cluster amplitudes (![]() ) needs to be evaluated instead of
two (one for the ground state and one for the energy difference):
) needs to be evaluated instead of
two (one for the ground state and one for the energy difference):
The following is an example for the CC2 single point calculation for an an excited state gradient (not that in the present implementation it is not possible to compute gradients for several excited states at the same time):
$ricc2 cc2 $excitations irrep=a1 nexc=2 exprop states=all operators=diplen,qudlen xgrad states=(a1 2)A different input is again required for geometry optimizations: in this case the model and excited state for which the geometry should be optimized have to be specified in the data group
$ricc2
with the keyword geoopt:
$ricc2 geoopt model=cc2 state=(a1 2) $excitations irrep=a1 nexc=2 exprop states=all operators=diplen,qudlen