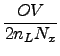



Next: Restrictions:
Up: Second-order Møller-Plesset Perturbation Theory
Previous: RI-MP2-F12 Calculations
Contents
Index
Laplace-transformed SOS-RI-MP2 with
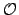 (
(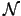 4) scaling costs
4) scaling costs
The ricc2 module contains since release 6.1 a first implementation of
SOS-MP2 which exploits
the RI approximation and a Laplace transformation of the orbital energy denominators
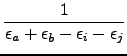 = = 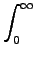 e-(εa+εb-εi-εj)tdt e-(εa+εb-εi-εj)tdt 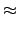 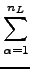 wαe-(εa+εb-εi-εj)tα , wαe-(εa+εb-εi-εj)tα , |
(8.8) |
to achieve an implementation with
(4) scaling costs, opposed to
the conventional
(5) scaling implementation.
In particular for large molecules the Laplace-transformed implementation can reduce
a lot the computational costs of SOS-MP2 calculations without loss in accuracy.
The Laplace-transformed implementation for SOS-MP2 calculations
is activated with the input
$laplace
conv=5
where the parameter conv is a convergence threshold for the numerical integration
in Eq. (8.8). A value of conv=5 means that the
numerical integration will be converged to a root mean squared error of
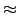 10-5 a.u.
10-5 a.u.
Whether the conventional or the Laplace-transformed implementation will be more
efficient depends firstly on the system size (the number of occupied orbitals) and
secondly on the required accuracy (the number of grid points for the numerical integration
in Eq. (8.8)) and can be understood and estimated from
the following considerations:
- The computational costs for the most expensive step in (canonical)
RI-MP2 energy calculations for large molecules requires
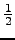 O2V2Nx floating point multiplications, where
O and V are, respectively, the number occupied and virtual orbitals and
Nx is the number of auxiliary functions for the RI approximation.
For the LT-SOS-RI-MP2 implementation the most expensive step involves
nLOVNx2 floating point multiplications, where nL is the number
of grid points for the numerical integration. Thus, the ratio of the computational
costs is approximately
where for the last step
Nx 3V has been assumed.
Thus, the Laplace-transformed implementation will be faster than
the conventional implementation if O > 6nL.
O2V2Nx floating point multiplications, where
O and V are, respectively, the number occupied and virtual orbitals and
Nx is the number of auxiliary functions for the RI approximation.
For the LT-SOS-RI-MP2 implementation the most expensive step involves
nLOVNx2 floating point multiplications, where nL is the number
of grid points for the numerical integration. Thus, the ratio of the computational
costs is approximately
where for the last step
Nx 3V has been assumed.
Thus, the Laplace-transformed implementation will be faster than
the conventional implementation if O > 6nL.
The number of grid points nL depends on the requested accuracy and the
spread of the orbital energy denominators in Eq. (8.8).
The efficiency of Laplace-transformed SOS-RI-MP2 calculations can
therefore (in difference to conventional RI-MP2 calculations) be enhanced significantly
by a carefull choice of the thresholds, the basis set, and the orbitals included
in the correlation treatment:
- The threshold
conv for the numerical integration
is by default set to the value of conv specified for
the ground state energy in the data group $ricc2 (see Sec. 15.2.13),
which is initialized using the threshold $denconv, which
by default is set conservatively to the tight value of 10-7.
- For single point energy calculations
conv in $laplace can
savely be set to 4, which gives SOS-MP2 energies converged within
10-4 a.u. with computational costs reduced by one third or more compared to
calculations with the default settings for these thresholds.
- For geometry optimizations with SOS-MP2 we recommend to set
conv in
$laplace to 5.
- The spread of the orbital energy denominators depends on the basis sets and the
orbitals included in the correlation treatment.
Most segmented contracted basis sets of triple-ζ or higher accuracy
(as e.g. the TZVPP and QZVPP basis sets) lead to rather high lying
``anti core'' orbitals with orbital energies of
10 a.u. and more.
- For the calculation of SOS-MP2 valence correlation energies
it is recommended to exclude such orbitals from the correlation treatment
(see input for $freeze in Sec. 15).
- Alternatively one can use general contracted basis sets, as e.g. the correlation consistent cc-pVXZ basis sets.
But note that general contracted basis sets
increase the computational costs for the integral evaluation in the
Hartree-Fock and, for gradient calculations, also the CPHF equations
and related 4-index integral derivatives.
- Also for the calculation of all-electron correlation energies
with core-valence basis sets which include uncontracted steep functions
it is recommended to check if extremely high-lying anti core orbitals
can be excluded.
Note that for large molecules it is recommended to disable for geometry optimizations
(or for gradient or property calculations in general) the preoptimization for the
Z vector equations with the nozpreopt option in the $response
data group (see Sec. 15.2.13).
Subsections
Next: Restrictions:
Up: Second-order Møller-Plesset Perturbation Theory
Previous: RI-MP2-F12 Calculations
Contents
Index
TURBOMOLE
 =
=