- $mvd
-
leads to calculation of relativistic corrections for the SCF total density in
case of dscf and ridft, for the SCF+MP2 density in case of rimp2 and
mpgrad and for that of the calculated excited state in case of egrad.
Quantities calculated are expectation values
< p2 > , < p4 > and the Darwin
term (
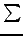 1/ZA*ρ(RA)).
1/ZA*ρ(RA)).
- $moments
-
yields calculation of electrostatic moments arising from nuclear charges and
total electron densities. Also without setting this keyword moments up to
quadrupole are calculated, with respect to reference point (0,0,0).
Supported extensions:
$moments <i>
x1 y1 z1
x2 y2 z2
.
.
By integer i; the maximum order of moments is specified, maximum and
default is i=3 (octopole moments), real numbers x, y, z allow for the
specification of one or more reference points.
- $pop
-
drives the options for population analyses. By default a Mulliken PA in the
basis of cartesian atomic orbitals (CAOs) is performed
for the total density (
Dα + Dβ) leading to Mulliken (brutto)
charges and, in case of spin-unrestricted calculations also for the spin
density (
Dα - Dβ) leading to Mulliken (brutto) numbers for
unpaired electrons. Besides total numbers also contributions from s-, p-,
...functions are listed separately.
Two-component wavefunctions (only module ridft and only if $soghf is set):
In two-component calculations instead of Sz
|(Sx, Sy, Sz)| is written to the output.
Additionally a vector-file named
spinvec.txt is written, which includes the resulting spinvector
for each atom in the system (also the direction).
The following modifications and extensions are supported, if the respective
commands are written in
the same line as $pop:
- lall
- Additional information about
px, py, pz (and analogous
for d and f functions) is displayed (lengthy output).
- atoms list of atoms
-
Contributions are plotted only
if arising
from atoms selected by list.
- thrpl=real
-
Contributions smaller than thrpl
are not
displayed (default: 0.01).
- overlap
- Mulliken atomic overlap matrix is displayed.
- netto
- Mulliken netto populations (diagonal elements of Mulliken
overlap matrix) are calculated.
- mosum list of MOs
-
Summed Mulliken contributions for
a group
of molecular orbitals defined by numbers referring to the numbering
obtained e.g. from the tool eiger. Note that occupancy of MOs is
ignored, i.e. all orbitals are treated as occupied.
- mo list of MOs
-
Mulliken contributions for single
MOs
defined by numbers (independent of whether they are occupied or not).
If this option is valid, one may additionally set
- dos width=real
points=integer
-
to calculate a (simulated) density of states by broadening the
discrete energy levels with Gaussians and superimposing them.
The width of each Gaussian may be set by input (default:
0.01a.u.). The resolution (number of points) may be chosen
automatically (default values are usually sufficient to generate
a satisfactory plot) or specified by hand. The output files
(dos in case of RHF wave functions, and
dos_a+b, dos_a-b,
dos_alpha, dos_beta; for UHF cases)
contain energies (first column), resulting DOS for the respective
energy (second column) as well as s-, p-, d-contributions
for the respective energy (following columns).
Example:
$pop mo 23-33 dos atoms 2,3,7-8
leads to Mulliken PA (CAO-basis) for each of the eleven MOs 23-33,
regarding only contributions from
atoms 2-3 and 7-8 (results are written to standard output) and
generation of file(s) with the
respective simulated density of states.
- $pop nbo
-
to perform a natural population analyses [121].
The possible options (specified in the same line) are
idbgl=integer Debug level.
AOmust be provided, the CAO case is notimplemented.
tw=realThreshold tw to circumvent numericaldifficulties incomputing Ow
(default: tw=1.d-6).
idbgl=integer Debug level
(default: idbgl=0).
abFor UHF cases: Print alpha and beta density results.
shortPrint only natural electron configuration and summary.
Example:
$pop nbo AO ab short atoms 1,2,6
leads to a natural population analysis (AO-basis) with printing the results of
alpha
and beta densities (only the electron configuration and the summary) for the
atoms 1,2 and 6.
To change the NMB set for atoms, one has to add a $nbonmb-block in the
control
file. Example:
$nbonmb
ni s:4 p:2 d:1
o s:2 p:1
leads to a NMB set for Ni of 4 s-, 2 p- and 1d-functions and for O of 2 s- and 1
p-functions.
- $pop paboon
-
to perform a population analyses based on occupation numbers
[122] yielding "shared electron numbers (SENs)" and
multicenter contributions. For this method always the total density is used,
i.e. the sum of alpha and beta densities in case of UHF, the SCF+MP2-density
in case of MP2 and the GHF total density for (two-component-)GHF.
The results of such an analysis may depend on the choice of the number of
modified atomic orbitals ("MAOs"), which can be specified by an additional
line; without further specification their number is calculated by the method
"mix", see below. Note: One should carefully read the information concerning
MAOs given in the output before looking at the numbers for atomic charges and
shared electron numbers.
- $mao selection options
-
to specify how MAOs are selected per atom.
Available options are:
a) for the way of sorting MAOs of each atom:
- eig
-
MAOs are sorted according to their eigenvalue (those with largest EW finally
are chosen). This is the default.
- occ
-
MAOs are sorted according to their occupation; note that the number of all
occupation is NOT the number of electrons in the system. This option is kept
rather for historical reasons.
b) for the determination of the number of MAOs:
- fix
-
A fixed number of MAOs is taken for each atom; usually this is the number of
shells up to the complete valence shell, e.g. 5 for B-Ne, 6 for Na-Mg, etc.
Exceptions are Elements Sc (Y, La), Ti (Zr, Hf), V (Nb, Ta) for which not
all five d-shells are included, but only 2, 3 or 4, respectively. This
modification leads to beetter agreement with partial charges calculated by
an ESP-fit.
- thr <real>
-
All MAOs with an eigenvalue larger than <real> are chosen; default
is <real>=0.1. This and the following two options are not valid in
connection with occ.
- max
-
Maximum of numbers calculated from fix and thr=0.1 is taken.
- mix
-
2:1 mixture of fix and thr=0.1. This choice gives best
agreement (statistical) with charges from an ESP-fit. It is the default
choice.
c) for additional information about MAOs:
- info
-
Eigenvalues and occupations for each MAO are written to output.
- dump
-
Entire information about each MAO is written to output. Lenghty.
Further for each atom the number of MAOs and the sorting mode can be
specified individually in lines below this keyword. Example:
atom 1,3-4 eig 7
leads to choice of the 7 MAOs with largest eigenvalue at atoms 1, 3-4.
- $localize
-
enables the generation of localized molecular orbitals (LMOs) using Boys
localization. By default,
all occupied orbitals are included, localised orbitals are written (by default
in the AO-basis) to
file(s) lmo in case of RHF and lalp and
lbet in case of UHF orbitals. Note,
that LMOs usually break the molecular symmetry; so, even for symmetric cases
the AO (not the SAO)
basis is used for the output. The localized orbitals are sorted with respect
to the corresponding
diagonal element of the Fock matrix in the LMO basis. In order to characterize
these orbitals,
dominant contributions of (canonical) MOs are written to standard output as
well as results of a
Mulliken PA for each LMO (for plotting of LMOs see option $pointval).
The keyword allows for following options (to be written in the same line):
- mo list of MOs
-
Include only selected MOs (e.g. valence MOs) in
localization procedure (numbering as available from
Eiger).
- sweeps=integer
-
maximum number of orbital rotations
to get LMOs;
default value is 10000 (sometimes not enough, in particular for highly
delocalised
systems).
- thrcont=real
-
lower threshold for displaying MO
and Mulliken
contributions (default: 0.1).
- CAO
- LMOs are written to file in the CAO basis (instead of AO)
- $esp_fit
-
fits point charges at the positions of nuclei to electrostatic potential
arising from electric
charge distribution (also possible for two-component calculations, for UHF
cases also for spin density). For this purpose the ("real")
electrostatic potential is calculated at spherical shells of grid points
around the atoms. By
default, Bragg-Slater radii, rBS, are taken as shell radii, for each atom
the number of
points is given by
1000⋅rBS2, the total number of points is
the sum of points for each
atom reduced by the number of points of overlapping spheres. Non-default
shells (one or more) can
be specified as follows:
$esp_fit
shell i1 s1
shell i2 s2
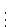
Integer numbers i define the number of points for the respective
shell, real numbers
s constants added to radii (default corresponds to one shell with s=1.0).
A parametrization very close to that by Kollman (U.C. Singh, P.A. Kollman, J. Comput. Chem.
5(2), 129-145 (1984)) may be obtained by
$esp_fit kollman
Here five shells are placed around each atom with r=1.4*rvdW + k,
k=0pm, 20pm, 40pm, 60pm, 80pm,
and rvdW are the van-der-Waals radii of the atoms.
- $pointval
-
drives the calculation of space-dependent molecular quantities at 3D grids,
planes, lines or
single points. Without further specifications the values of densities are
plotted on a
three-dimensional grid adapted to the molecular size. Data are deposed to
output files (suffix
plt) that can be visualized directly with the gOpenMol program.
In case of RHF-dscf/ridft
calculations you get the total density on file td.plt, for
UHF-dscf/ridft calculations one
gets both values for the total density (
Dα + Dβ) on
td.plt and the "spin
density" (
Dα - Dβ) on sd.plt. For mpgrad/rimp2
calculations one gets in the
RHF case the total density (D(SCF+MP2)) on td.plt and the MP2
contribution on
mp2d.plt and in the UHF case one obtains the total density
(
Dα(SCF + MP2) + Dβ(SCF + MP2)) on td.plt, the "spin density"
(
Dα(SCF + MP2) - Dβ(SCF + MP2)) on td.plt, and the
respective MP2 contributions
on files mp2d.plt and mp2sd.plt. For egrad it is similar,
just replace in the
filenames mp2 by e.
Integration of density (if absolute value greater than eps) within a
sphere (origin x, y, z, radius r) is performed for
- $pointval integrate x y z r eps
-
By default the origin is at (0,0,0), the radius is chosen large enough to
include the whole 3D box and all contributions are regarded (eps=0).
Data different from total and spin densities are generated by following
(combinable) settings
(to be written in the same line as statement $pointval):
- pot
- leads to calculation of electrostatic potential arising
from electron densities, nuclei and--if present--constant electric fields and
point charges. The densities used for calculation of potentials are the
same as above; the respective filenames are generated from those of
densities by replacement of the "d" (for density) by a "p" (for potential).
By "
pot eonly" only the electronic contribution to the electrostatic
potential is calculated.
- fld
- calculation of electric field. Note, that for 3D default
output format (.plt, see below) only norm is displayed. Densities used
are the same as above, filenames are generated from those of densities
by replacement of "d" (for density) by "f" (for field).
- mo list of MO numbers
-
calculation of amplitudes
of MOs specified by numbers referring to the numbering
obtained e.g. from the tool eiger in the same format. The respective
filenames are self-explanatory and displayed in the output. Note, that
also in MP2 and excited state calculations the HF/DFT ground state orbitals
are plotted (and not natural MP2/excited orbitals).
Two-component cases: The density of the spinors specified by numbers referring
to the numbering obtained e.g. from the file EIGS are visualized. By
setting the keyword minco also the amplitudes of the spinor-parts are
calculated, whose weights (the probability of finding the electron in this part)
lie above the threshold.
- lmo list of LMO numbers
-
calculation of amplitudes
of LMOs
(previously generated by $localize) ordered by the corresponding
diagonal element of the Fock matrix in the LMO basis.
- nmo list of NMO numbers
-
calculation of amplitudes
of NMOs
(previously generated by
$natural orbitals file=natural and
$natural orbital occupation file=natural
- dens
- has to be set, if additionally to one of the above
quantities also the density is to be computed.
- xc
- calculation of the Kohn-Sham exchange-correlation potential.
It is only valid for DFT calculations and it works for all exchange-correlation
functionals, including LHF.
Note that for hybrid functionals, only the Kohn-Sham part of the potential will
be computed (the HF part is a non-local-operator and can't be plotted).
For GGA/MGGA functional only the local part is (currently) computed.
For line plots the output file is tx.vec .
For UHF calculatiosn the output files are
tx.vec (alpha-spin potential) and sx.vec (beta-spin potential).
For line plot the files has three columns:
1: total potential
2: local part ( or Slater-potential for LHF)
3: non-local part ( or Correction term for LHF)
Output formats may be specified by e.g. fmt=xyz if written to the
same line as $pointval. Supported are:
- xyz
- in case of scalars (density, (L)MO amplitudes,
electrostatic potential) this format is:
(x, y, z, f (x, y, z)). In case of
vectors components of the vector and its norm are displayed. This format
is valid for all types of grid (3D, plane, line, points, see below),
it is the default format in case of calculation of values at single
points. Output file suffix is
.xyz.
- plt
- only for 3D, default in this case. Data are written to
binary files that can be directly read by gOpenMol. Note, that this output
is restricted to scalar quantities; thus in case of vectors (E-field) only
the norm is plotted. Output file suffix is
.plt.
- map
- only for 3D. Data are written to ASCII files that
can be imported by e.g. gOpenMol. Note, that this output is restricted to
scalar quantities; thus in case of vectors (E-field) only the norm is plotted.
Output file suffix is .map.
- txt
- a format compatible with gOpenMol for visualization of vectors v.
The format is
x, y, z, vx, vy, vz.
- vec
- for planes and lines (default in these cases). In case of
a line specified by
α⋅
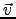 (see below) output is
α, f (x, y, z) for scalars, for vectors components and norm are displayed.
vectors. Analogously, in case of planes it is
α, β, f (x, y, z). The output (file suffix
(see below) output is
α, f (x, y, z) for scalars, for vectors components and norm are displayed.
vectors. Analogously, in case of planes it is
α, β, f (x, y, z). The output (file suffix .vec) may be visualized by plotting
programs suited for two-dimensional plots. A command
file (termed gnuset) to get a contour plot by gnuplot is automatically
generated.
- cub
- only for 3D, writes out data in Cube format which can be imported by
many external visualization programs.
For 3D grids non-default boundarys, basis vector directions,
origin and resolutions may be specified as follows:
$pointval
grid1 vector 0 3 0 range -2,2 points 200
grid2 vector 0 0 -7 range -1,4 points 300
grid3 vector 1 0 0 range -1,1 points 300
origin 1 1 1
Grid vectors (automatically normalised) now are (0, 1, 0),(0, 0, - 1),(1, 0, 0),
the grid is centered at (1, 1, 1), and e.g. for the first direction 200 points
are distributed between -2 and 2.
Grids of lower dimensionality may be specified (in the same line as
$pointval) by typing either geo=plane or geo=line
or geo=point The way to use is best explained by some examples:
$pointval geo=plane
grid1 vector 0 1 0 range -2,2 points 200
grid2 vector 0 0 1 range -1,4 points 300
origin 1 1 1
Values are calculated at a plane spanned by vectors (0,1,0) and (0,0,1) centered
at (1,1,1).
$pointval geo=line
grid1 vector 0 1 0 range -2,2 points 50
origin 0 0 1
Values are calculated at a line in direction (0,1,0) centered
at (0,0,1). Output format as above.
$pointval geo=point
7 5 3
0 0 7
Values are calculated at the two points
(7.0, 5.0, 3.0) and
(0.0, 0.0, 7.0).