



Next: Isodensity Cavity:
Up: Format of Keywords and
Previous: Keywords for Periodic Electrostatic
Contents
Index
Keywords for COSMO
The Conductor-like Screening Model (cosmo) is a
continuum solvation model, where the solute molecule forms
a cavity within the dielectric continuum of permittivity epsilon
that represents the solvent. A brief description of the method is
given in chapter 14.
The model is currently implemented for SCF energy and gradient calculations
(dscf/ridft and grad/rdgrad), MP2 energy calculations (RIMP2 and
mpgrad) and MP2 gradients (RIMP2).
For simple HF or DFT single point calculations or optimizations with standard
settings, we recommend to add the $cosmo
keyword to the control file and to skip the rest of this section.
Please note: due to improvements in the 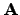 matrix and cavity setup the
cosmo energies and gradients may differ from older versions (5.7 and older). The
matrix and cavity setup the
cosmo energies and gradients may differ from older versions (5.7 and older). The
use_old_amat option can be used to calculate energies (not gradients)
using the old cavity algorithm of TURBOMOLE 5.7.
The basic cosmo settings are defined in the $cosmo and the
$cosmo_atoms block.
Example with default values:
$cosmo
epsilon=infinity
nppa= 1082
nspa= 92
disex= 10.0000
rsolv= 1.30
routf= 0.85
cavity closed
ampran= 0.1D-04
phsran= 0.0
# the following options are not used by default
allocate_nps= 140
use_old_amat
no_oc
- epsilon=real
-
defines a finite permittivity used for
scaling the screening charges.
- allocate_nps=integer
-
skips the cosmo segment
statistics run and allocates memory for the given number of segments.
- no_oc
-
skips the outlying charge correction.
All other parameters affect the generation of the surface
and the construction of the
A matrix:
- nppa= integer
-
number of basis grid points per
atom
(allowed values:
k = 10×3i×n2 + 2 = 12, 32, 42, 92...)
- nspa= integer
-
number of segments per atom
(allowed values:
k = 10×3i×n2 + 2 = 12, 32, 42, 92...)
- disex= real
-
distance threshold for A matrix
elements (Ångstrom)
- rsolv= real
-
distance to outer solvent sphere
for cavity
construction (Ångstrom)
- routf= real
-
factor for outer cavity
construction in the outlying
charge correction
- cavity closed
-
fill in seams between atoms with points
- cavity open
-
leave untidy seams between atoms
- ampran= real
-
amplitude of the cavity
de-symmetrization
- phsran= real
-
phase of the cavity
de-symmetrization
- use_old_amat
-
uses matrix setup of TURBOMOLE 5.7
If the $cosmo keyword is given without further specifications the
default parameter are used (recommended).
For the generation of the cavity, cosmo also requires the definition
of atomic radii. User defined values can be provided in Ångstrom units in the
data group $cosmo_atoms, e.g. for a water molecule:
$cosmo_atoms
# radii in Angstrom units
o 1 \
radius= 1.7200
h 2-3 \
radius= 1.3000
If this section is missing in the control file, the default
values defined in the radii.cosmo file (located in $TURBODIR/parameter) are used.
A user defined value supersedes this defaults.
$cosmo and $cosmo_atoms can be set interactively with the cosmo input program Cosmoprep after the usual generation of the
TURBOMOLE input.
The cosmo energies and total charges are listed in the result section. E.g.:
SCREENING CHARGE:
cosmo : -0.003925
correction : 0.003644
total : -0.000282
ENERGIES [a.u.]:
Total energy = -76.0296831863
Total energy + OC corr. = -76.0297567835
Dielectric energy = -0.0118029468
Diel. energy + OC corr. = -0.0118765440
The following value is included for downward compatibility
Total energy corrected = -76.0297199849
The dielectric energy of the system is already included in the total energy.
OC corr denotes the outlying charge correction. The last energy entry
gives the total outlying charge corrected energy in the old definition used in
TURBOMOLE 5.7 and older versions.
Subsections
Next: Isodensity Cavity:
Up: Format of Keywords and
Previous: Keywords for Periodic Electrostatic
Contents
Index
TURBOMOLE